

Forschende der Charité – Universitätsmedizin Berlin, des Max-Planck-Instituts für molekulare Genetik (MPIMG) und des Universitätsklinikums Schleswig-Holstein (UKSH) haben im Detail aufgeklärt, wie das äußerst seltene erbliche BPTA-Syndrom entsteht: Die Ladungsänderung eines Proteins bringt die zelluläre Selbstorganisation durcheinander, eine Entwicklungsstörung ist die Folge. Das Team identifizierte außerdem hunderte vergleichbare genetische Veränderungen, die unter anderem mit Störungen der Hirnentwicklung oder einer Krebs-Veranlagung in Zusammenhang stehen. Der jetzt im Fachmagazin Nature* beschriebene Mechanismus könnte damit die Ursache für zahlreiche unaufgeklärte Erkrankungen sein.
Tausende genetische Veränderungen stehen in Verbindung mit verschiedenen Erkrankungen. Wie genau diese Mutationen zu Krankheiten führen, ist meistens jedoch unklar. Das liegt daran, dass sie Abschnitte von Proteinen betreffen, deren dreidimensionale Struktur ungeordnet ist und über deren Funktion in der Zelle man bisher wenig weiß. „Die Aufgaben solcher Proteinabschnitte sind schwer zu erforschen, weil sie häufig erst zusammen mit anderen Molekülen ihre Wirkung entfalten", sagt Dr. Martin Mensah vom Institut für Medizinische Genetik und Humangenetik der Charité. Er ist einer der beiden Erstautoren der Studie und Fellow des Digital Clinician Scientist Programms, das die Charité zusammen mit dem Berlin Institute of Health in der Charité (BIH) betreibt. „Am Beispiel des BPTA-Syndroms haben wir nun im Detail beschrieben, wie Veränderungen in ungeordneten Proteinbereichen eine genetisch bedingte Krankheit verursachen können." Damit hat das Forschungsteam einen neuen Entstehungsmechanismus von Erbkrankheiten entdeckt – der der Studie zufolge überraschenderweise gar nicht so selten ist.
BPTA steht für „Brachyphalangie-Polydaktylie und tibiale Aplasie/Hypoplasie". Die Betroffenen haben schwerwiegende Fehlbildungen an den Gliedmaßen, dem Gesicht, dem Nerven- und Knochensystem und anderen Organen. Mit weniger als zehn dokumentierten Fällen weltweit ist die Krankheit extrem selten. Um der Ursache für das BPTA-Syndrom auf die Spur zu kommen, entschlüsselten die Forschenden die Erbinformation von fünf Betroffenen und stellten fest, dass bei allen das Protein HMGB1 verändert ist: Das letzte Drittel seiner Struktur ist durch eine sogenannte Rasterschub-Mutation nicht länger negativ, sondern positiv geladen.
Das Kernkörperchen verfestigt sich
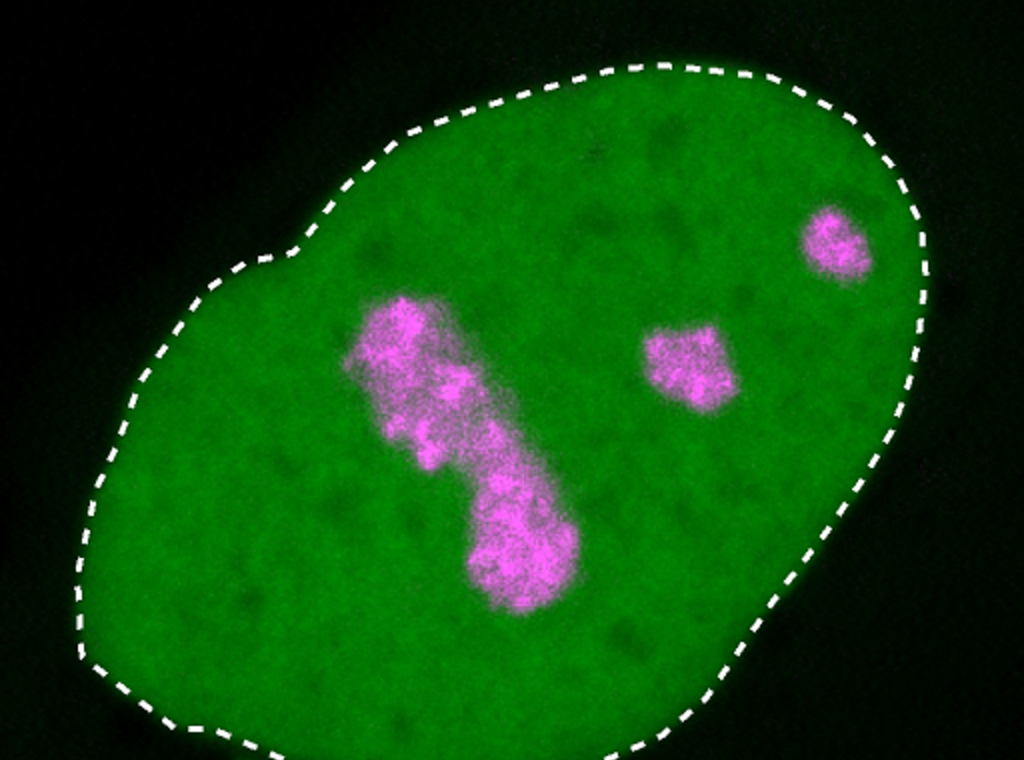
Durch die Ladungsänderung ähnelt HMGB1 nun Proteinen, die sich vorzugsweise im sogenannten Kernkörperchen aufhalten. Das Kernkörperchen ist ein kleiner Bereich im Zellkern, in dem Teile der Proteinfabriken zusammengebaut werden. Es ist daher fundamental wichtig für die Lebensfähigkeit einer Zelle. Wie das Forschungsteam anhand von Versuchen mit isolierten Proteinen und Zellkulturen belegte, wird das mutierte HMGB1-Protein mit seinem nun positiv geladenen Endstück fälschlicherweise zum Kernkörperchen hingezogen. Weil der Proteinfortsatz zum Teil auch zäher geworden ist, verklumpt das HMGB1-Protein außerdem. „Im Mikroskop konnten wir nachvollziehen, dass das Kernkörperchen dadurch seine eigentlich flüssigkeitsähnlichen Eigenschaften verliert und zunehmend erstarrt", erklärt Dr. Henri Niskanen, Wissenschaftler am MPIMG und ebenfalls Erstautor der Studie.
Die Verfestigung des Kernkörperchens beeinträchtigt die Lebensfunktion der Zellen: Mit dem mutierten Protein starben mehr Zellen in der Kultur als ohne die Mutation. Prof. Dr. Malte Spielmann, Direktor des Instituts für Humangenetik des UKSH und einer der drei leitenden Autoren der Studie, resümiert: „Wir haben also gezeigt, wie Mutationen in ungeordneten Proteinabschnitten eine Krankheit verursachen können: Durch eine Ladungsänderung sammelt sich das Protein fälschlicherweise im Kernkörperchen an und beeinträchtigt so dessen lebenswichtige Funktion. In der Folge ist die Entwicklung des Organismus gestört."
Existierende Krankheiten neu erklärt
Anschließend durchsuchte das Forschungsteam in Datenbanken die DNA-Sequenzen von tausenden Personen auf der Suche nach ähnlichen Fällen. Tatsächlich konnten die Wissenschaftlerinnen und Wissenschaftler mehr als 600 Mutationen in 66 Proteinen identifizieren, die dem Protein-Endstück sowohl eine positive Ladung als auch zähere Eigenschaften verliehen. 101 davon waren bereits zuvor mit verschiedenen Krankheiten in Verbindung gebracht worden, darunter neuronale Entwicklungsstörungen und eine erhöhte Anfälligkeit für Krebs. Für 13 ausgewählte Proteine prüfte das Team in der Zellkultur, ob sie durch die Mutationen eine Affinität für das Kernkörperchen erhielten. Das traf in zwölf Fällen zu. Etwa die Hälfte der getesteten Proteine beeinträchtigten die Funktion des Kernkörperchens und ähnelten damit dem Krankheitsmechanismus des BPTA-Syndroms.
„Der Entstehungsmechanismus, den wir beim BPTA-Syndrom entdeckt haben, könnte also bei vielen weiteren Krankheiten zum Tragen kommen", sagt Prof. Dr. Denise Horn, leitende Studienautorin vom Institut für Medizinische Genetik und Humangenetik der Charité. „Wir stoßen damit eine Tür auf, die zur Aufklärung zahlreicher weiterer Erkrankungen führen könnte. Die eigentliche Arbeit beginnt deshalb erst jetzt."
Zumindest für einige Erkrankungen könnte der jetzt bekannte Mechanismus außerdem einen neuen Therapieansatz liefern. „Tumorleiden sind auf genetische Veränderungen in den betroffenen Zellen zurückzuführen", erklärt Dr. Denes Hnisz, Forschungsgruppenleiter am MPIMG und dritter leitender Autor der Studie. „Möglicherweise können wir in Zukunft also die Krebsentwicklung unterbinden, indem wir in die Selbstorganisation der Zelle eingreifen, die über ungeordnete Proteinabschnitte vermittelt wird."


